
今回は、そういうお話です。
※Eも、高分解能 or 高溶媒含量の場合には、結構キレイな位相をつけるんですけどねぇ…
shelxDは、見つけた重原子位置をPDBフォーマットでファイル出力(*_fa.pdb)します。
CRYST1 78.500 78.500 78.500 90.00 90.00 90.00 SCALE1 0.012739 -0.000000 -0.000000 -0.00000 SCALE2 -0.000000 0.012739 -0.000000 0.00000 SCALE3 0.000000 -0.000000 0.012739 -0.00000 ATOM 1 S UNK 1 -20.219 -1.554 -12.937 1.00 20.00 ATOM 2 S UNK 2 -13.200 2.020 -5.879 0.75 20.00 ATOM 3 S UNK 3 -28.074 -5.902 -10.752 0.64 20.00 ATOM 4 S UNK 4 -12.519 3.034 -7.538 0.46 20.00 ATOM 5 S UNK 5 -27.973 -6.668 -11.783 0.41 20.00 ATOM 6 S UNK 6 -15.793 6.357 -5.934 0.40 20.00 ATOM 7 S UNK 7 -8.074 0.238 -17.357 0.35 20.00 ATOM 8 S UNK 8 -21.993 -10.205 -10.905 0.32 20.00 ATOM 9 S UNK 9 -28.777 -2.064 -7.107 0.27 20.00 END
これを shelxE 以外のソフトウェアで精密化したい場合には、適宜フォーマット変換しなければなりません。
例えば、ccp4 に含まれる mlphare で精密化する場合には、CCP4_HAフォーマットに変換します。この変換には「coordconv」というプログラムを用います( ccp4i なら、【Corrdinate Utilities】→【Convert Coordinate Formats】)。

変換された HAフォーマットは、フラクショナル表現となっています。
ATOM1 S -0.25757 -0.01980 -0.16480 1.00 0.0 BFAC 20.000 ATOM2 S -0.16815 0.02573 -0.07489 0.75 0.0 BFAC 20.000 ATOM3 S -0.35763 -0.07518 -0.13697 0.64 0.0 BFAC 20.000 ATOM4 S -0.15948 0.03865 -0.09603 0.46 0.0 BFAC 20.000 ATOM5 S -0.35634 -0.08494 -0.15010 0.41 0.0 BFAC 20.000 ATOM6 S -0.20118 0.08098 -0.07559 0.40 0.0 BFAC 20.000 ATOM7 S -0.10285 0.00303 -0.22111 0.35 0.0 BFAC 20.000 ATOM8 S -0.28017 -0.13000 -0.13892 0.32 0.0 BFAC 20.000 ATOM9 S -0.36659 -0.02629 -0.09054 0.27 0.0 BFAC 20.000
ただし、このままですと「Anormalous Occupancy」が0.0ですので、mlphare にかける場合には、これを全て1.0に書き直す必要があります(参考)。
精密化に Solve を用いる場合、重原子位置はパラメータとして与えることになります。
このフォーマットは、HA フォーマットと同様なフラクショナル表現なんですが、その記述順が若干異なっています(ここでは便宜的に「XYZフォーマット」と呼ぶ)。
… mad_atom S lambda 1 label SAD data for S wavelength 2.600 fprimv_mad 0.3357 fprprv_mad 1.4246 atomname S xyz -0.25757 -0.01980 -0.16480 occupancy 1.000 bvalue 20.000 xyz -0.16815 0.02573 -0.07489 occupancy 0.750 bvalue 20.000 xyz -0.35763 -0.07518 -0.13697 occupancy 0.640 bvalue 20.000 …
まぁ、1つ1つ手で書き換えてもイイんですが、面倒なので ccp4i タスク「ha2xyz」を作ってみました。
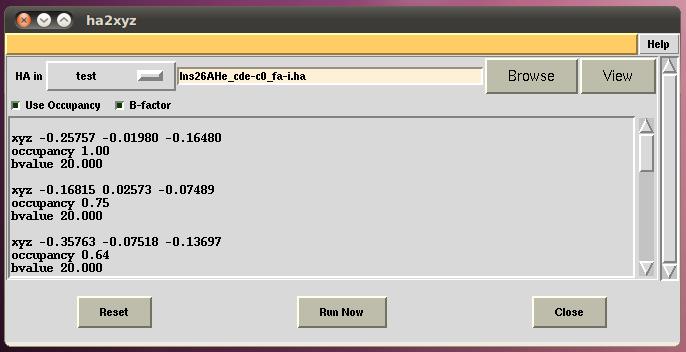
インストールすると、【Corrdinate Utilities】の一番下と、【Program List】のアルファベット順の2箇所に入ります。
【HA in】で指定したファイルを、XYZフォーマットに変換してテキストフレーム内に出力しますので、それを solve スクリプトにコピペすればOKです。
shelxD →Solve の場合には、「coordconv」→「ha2xyz」と2回操作がいるのが面倒ですが、我慢してくださいw
※ちなみに、以前アップしたコレだと、直接HAフォーマットを読み込めるようになってます
0 件のコメント:
コメントを投稿